Dealing with Pseudo-Replication in Linear Mixed Models
Pseudo-replication is associated with the incorrect identification of the number of samples or replicates in a study. This can occur at the planning or execution stage of a study, or during the statistical analyses. Almost always this incorrect identification will make the statistical analyses invalid.
To illustrate this, we will consider an experiment that compares two treatments applied to eight plots, each containing four plants (see figure below). In this experiment one treatment (_e.g._, control) is applied to four plots at random, and the other treatment (_e.g._, fertilizer) to the remaining four plots.
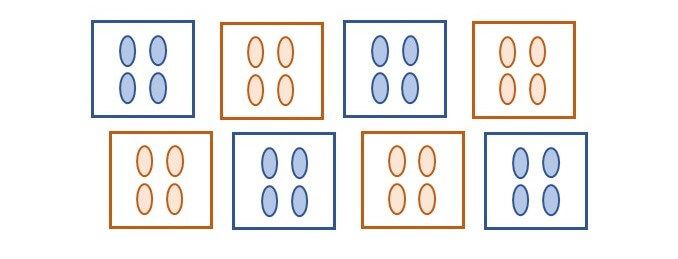
What is the replication of a given treatment in this experiment? The replication per treatment is four, but in total 16 plants are treated. The experimental units (EU) are the plots, while the individual plants, or measurement units (MU), are the pseudo-replicates. Even though we might have a total of 16 observations per treatment, these have a structure, and the treatment was randomized to the plots not to the plants.
In statistical terms if pseudo-replication is not properly accounted for the degrees of freedom (_df_) are incorrect, and generally inflated. This inflation of the _df_ leads to p-values that are lower than what they should be, and therefore, with pseudo-replication we are more likely to reject our null hypothesis. For example, in the experiment above, a t-test comparing the two treatment means should have a total of 6 _df_, but under pseudo-replication we will incorrectly consider that we have 30 _df_. In this case our critical t-value at a 5% significance level will change from 2.447 to 2.042, making our p-value and confidence interval incorrect. You can imagine, that pseudo-replication has important implications for statistical inference and scientific research, however, it is common to find this mistake in research papers.
In this topic, there are two important elements to consider:
1. _How to identify pseudo-replication in a study,_ and
2. _How to correctly deal with this pseudo-replication within a statistical analysis._
We will discuss both aspects with emphasis on the use of linear models (LM) and linear mixed models (LMM).
## Identifying pseudo-replication
Pseudo-replication is not always easy to recognize, particularly for experiments that contain multiple layers or strata. However, the most important aspect is to clearly distinguish between experimental units (EU) and the measurement units (MU). Treatment levels should always be applied randomly to different EU, as in the above example where a treatment level (_e.g._, fertilizer) was applied to a plot, and thus all four plants within the plot have the same treatment level. The difficulty comes when we take measurements (_e.g._, plant height) from each of the plants, and therefore the EU differs from the MU.
You can identify similar cases, for example where we obtain several measurements from a single EU, or even several measurements from a given MU. For example, in our experiment we could have counted insects in a sample of two leaves per plant. Here, these leaves will also be MU, but in this case, they correspond to a subsampling within each plant.
Careful identification of the design structure of an experiment, or equivalently of the arrangement (or layers) of the observations will allow us to identify the existence of pseudo-replication, and also it will help us to correctly specify this multi-stratum structure into a statistical model.
## Dealing with pseudo-replication
Linear mixed models (LMM) are particularly useful in dealing with the multi-stratum structure of an experiment, as they will recognize these layers and identify at what stratum there is pseudo-replication or true replication. Once the design and treatment structure are recognized the correct analysis follows easily. Further details on these structures and multi-stratum Analysis of Variance can be found in Welham _et al._ (2014).
For example, we will consider a dataset from a _Eucalyptus_ hybrid study. The original study evaluated clones from several intra- and inter-specific crosses between different _Eucalyptus_ species and their hybrids (Alves _et al._ 2018). Here, we will focus only on the hybrids between two species (_Eucalyptus urophylla_ and _E. resinifera_) corresponding to a total of 17 crosses or families. The study had a randomized complete block design (RCBD) with eight blocks, where propagules (_i.e._, trees) were arranged by family in plots of six plants. Hence, there is a total of 136 plots (8 blocks x 17 families) and 816 plants (136 plots each with 6 plants) in the dataset.
For this experiment, we can identify the structure (or stratum) as having replicates (or blocks) that are partitioned into several plots, and finally, we have individual trees within these plots. Hence, similar to the previous example, the plots are our EU and the trees are the MU. Under this structure the treatments (_i.e._, families) were assigned at the plot level and not at the plant level.
The data for our analyses can be found in the dataset called $\\texttt{EUCAL}$ and the first few lines of this data are:
| id | rep | plot | tree | family | male | female | height | dbh | volume |
| --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| 1 | 1 | 181 | 1 | 18 | 14 | 3 | NA | NA | NA |
| 2 | 1 | 181 | 2 | 18 | 14 | 3 | 25.4 | 16.39 | 0.2680 |
| 3 | 1 | 181 | 3 | 18 | 14 | 3 | 26.5 | 17.86 | 0.3318 |
| 4 | 1 | 181 | 4 | 18 | 14 | 3 | 20.6 | 17.57 | 0.2498 |
| 5 | 1 | 181 | 5 | 18 | 14 | 3 | 27.8 | 18.46 | 0.3721 |
| 6 | 1 | 181 | 6 | 18 | 14 | 3 | 20.5 | 11.62 | 0.1087 |
| 7 | 1 | 281 | 1 | 28 | 11 | 4 | NA | NA | NA |
| 8 | 1 | 281 | 2 | 28 | 11 | 4 | 26.1 | 18.18 | 0.3386 |
We observe that the first six observations correspond to plot 181 from replicate 1 However, this plot has only five trees alive at the time of measurement (data for tree 1 is missing, denoted with $\\texttt{NA}$). Hence, as with many experiments, we have a few missing observations.
Now, we can proceed to write the correct model that considers the full structure of this experiment:
$$
y\_{ijk} = \\mu + r\_i + f\_j + p\_{ij} + \\epsilon\_{ijk}
$$
where $y{ijk}$ is the phenotypic observation of the kth plant from the jth family on the ith replicate (or block); $\\mu$ is the overall mean; $ri$ is the fixed effect of the ith replicate; $f\_j$ is the fixed effect of the jth family; $p\_{ij}$ is random effect of the ijth plot, with $p\_{ij} \\sim N(0, \\sigma^2\_p)$; and $\\epsilon\_{ijk}$ is the random residual with $\\epsilon\_{ijk} \\sim N(0, \\sigma^2\_e)$.
In the above model we have defined the replicate and family terms as fixed effects. But depending on the objective of the analyses these could be considered random effects. The plot effect, $p\_{ij}$, was assumed to be random, because it is used here to describe the design structure of the study. The above model is easily fitted in much of the statistical software that will fit LMM. In this example, we will use ASReml-R as implemented in R.
## Fitting a LM to plant-level data
To demonstrate the effects of pseudo-replication in this experiment, we will be evaluating a few different models. First, we fit an incorrect model: one that ignores the plot effects, and therefore has pseudo-replication. The code for this model in ASReml-R is:
```plaintext
modINC<-asreml(fixed=height~rep+family,
data=EUCAL)
```
Recall that both $\\texttt{rep}$ and $\\texttt{family}$ are factors considered fixed effects. Our dataset called $\\texttt{EUCAL}$ has a row for each tree, and therefore a total of 816 records. Here, we are interested in the ANOVA table and the predicted means for the 17 families. These can be obtained using the commands:
```plaintext
wald.asreml(modINC,denDF='numeric')$Wald
predsINC<-predict(modINC,classify='family')$pvals
```
resulting in the ANOVA table:
| | Df | denDF | F.inc | Pr |
| --- | --- | --- | --- | --- |
| (Intercept) | 1 | 686 | 20280.000 | 0.0000000 |
| rep | 7 | 686 | 1.062 | 0.3864759 |
| family | 16 | 686 | 2.237 | 0.0036815 |
The predicted means for the first six families are:
| family | predicted.value | std.error | status |
| --- | --- | --- | --- |
| 18 | 24.06383 | 0.6799762 | Estimable |
| 28 | 25.72138 | 0.7499339 | Estimable |
| 102 | 23.48852 | 0.7052685 | Estimable |
| 104 | 22.91479 | 0.7311813 | Estimable |
| 158 | 26.25878 | 0.6799763 | Estimable |
| 172 | 24.60833 | 0.6582289 | Estimable |
In our analysis, we obtained an estimate of the residual variance, $\\hat{\\sigma}^2\_e$ of 20.80 (not shown), and the ANOVA table has 686 denominator _df_ for each fixed term. To better understand this ANOVA table we can write it as:
| Source | df | |
| --- | --- | --- |
| Replicate | r – 1 | \= 7 |
| Family | f – 1 | \= 16 |
| Error | r x f x m – r – f + 1 | \= 792 |
| Total | r x f x m -1 | \= 815 |
where r = 8 is the number of replicates, f = 17 is the number of families, and m = 6 is the number of plants per plot. The significance of replicate and family are assessed using the F-values of $F\_{7,792}$ and $F\_{16,792}$, respectively. Therefore, we have 792 _df_ for the denominator for these tests for any confidence interval we might calculate and for any contrast or multiple comparison we might desire to construct from this analysis.
The reason why our ANOVA table from ASReml-R has only 686 _df_ is because of the missing values in the dataset. For the response height, there are 106 missing values: 792 – 106 = 686.
The ANOVA denominator _df_, 686, is inconsistent with the total number of plots in the experiment: f x r = 136, a much smaller number.
## Fitting a LM to plot-level data
Probably one of the easiest ways to deal with multi-stratum data that avoids complex models, but that still fits a correct model, is to collapse the data by obtaining means, and then use these means as our response variable instead of the individual observations. The dataset called $\\texttt{EG\_PLOT}$, has a total of 136 observations (_i.e._, plots), and the first few lines of this data are:
| id | plot | rep | family | male | female | height | dbh | volume |
| --- | --- | --- | --- | --- | --- | --- | --- | --- |
| 1 | 181 | 1 | 18 | 14 | 3 | 24.1600 | 16.3800 | 0.2661 |
| 2 | 281 | 1 | 28 | 11 | 4 | 24.1600 | 15.1160 | 0.2388 |
| 3 | 1021 | 1 | 102 | 13 | 9 | 22.8167 | 13.7450 | 0.1912 |
| 4 | 1041 | 1 | 104 | 14 | 9 | 21.7667 | 13.2533 | 0.1785 |
| 5 | 1581 | 1 | 158 | 14 | 14 | 24.7500 | 15.2267 | 0.2374 |
| 6 | 1721 | 1 | 172 | 13 | 15 | 24.7667 | 16.0083 | 0.2535 |
| 7 | 1931 | 1 | 193 | 14 | 16 | 21.7750 | 13.2775 | 0.1900 |
| 8 | 1991 | 1 | 199 | 11 | 16 | 25.3000 | 15.9567 | 0.2631 |
The first observation for height corresponds to plot 181 from replicate 1, with a value of 24.16 m. This is the arithmetic average of five trees (recall that there is one dead plant for this plot).
Now, we are ready to fit the correct model to the plot means using the following code:
```plaintext
modPLOT<-asreml(fixed=height~rep+family,
data=EG_PLOT)
```
The above model does not contain any random effects, as plot is now absorbed by the residual term. That is, the residual term represents the error associated with the plot mean for height. Before, we look into the output, a generic ANOVA table for this model is:
| Source | df | |
| --- | --- | --- |
| Replicate | r – 1 | \= 7 |
| Family | f – 1 | \= 16 |
| Error | (r – 1) x (f – 1) | \= 112 |
| Total | r x f – 1 | \= 135 |
Here, we have that the significance of the replicate and family factors are assessed with F-values of $F\_{7,112}$ and $F\_{16,112}$, respectively. Hence, the denominator _df_ here is 112 instead of 792 (or 686 due to missing values) from the previous model. As you can expect, this reduction in _df_ will have important consequences. We can see this from requesting from ASReml-R the ANOVA table and the predicted means:
| | Df | denDF | F.inc | Pr |
| --- | --- | --- | --- | --- |
| (Intercept) | 1 | 686 | 20280.000 | 0.0000000 |
| rep | 7 | 686 | 1.062 | 0.3864759 |
| family | 16 | 686 | 2.237 | 0.0036815 |
The predicted means for the first six families are:
| family | predicted.value | std.error | status |
| --- | --- | --- | --- |
| 18 | 24.05501 | 0.8324633 | Estimable |
| 28 | 25.77875 | 0.8324633 | Estimable |
| 102 | 23.08334 | 0.8324633 | Estimable |
| 104 | 22.99771 | 0.8324633 | Estimable |
| 158 | 26.27583 | 0.8324633 | Estimable |
| 172 | 24.60834 | 0.8324633 | Estimable |
The ANOVA table from ASReml-R provides us with the correct _df_, and we can see that the significance of the family factor has a p-value of 0.0433 for this mean model analysis, in contrast with the p-value of 0.0037 for the incorrect model. If we are using a significance level of 1%, then our conclusions will be quite different! This is a clear consequence of pseudo-replication, where we have arrived at a different conclusion due to our incorrect consideration of the true-replication of an experiment.
The predicted values for the means in both analyses are very similar, with differences only due to how the missing data were treated (more on this below). But, the standard errors of the mean (SEM) from this analysis are 0.83, a value larger than the ones obtained earlier that ranged from 0.66 to 0.81; again, this is due to the fact that we have a different number of replications considered between the analyses.
## Fitting a LMM to plant-level data
Having identified the differences between an incorrect model that uses the plant-level data against a correct model that uses the plot-level data, now we are in a position to fit the correct model to the plant-level data based on our generic model defined before:
$$
y\_{ijk} = \\mu + r\_i + f\_j + p\_{ij} + \\epsilon\_{ijk}
$$
and this can be fitted with the code:
```plaintext
modCC<-asreml(fixed=height~rep+family,
random=~rep:plot,
data=EUCAL)
```
Here, we are specifying the plot term to be random, but this is written as $\\texttt{rep:plot} to ensure that each plot is uniquely identified (for example, we want to differentiate between plot 1 – replicate 1 from plot 1 – replicate 2).
As before, we will have a look at the generic ANOVA table for this model:
| Source | df | |
| --- | --- | --- |
| Replicate | r – 1 | \= 7 |
| Family | f – 1 | \= 16 |
| Plot | (r – 1) x (f – 1) | \= 112 |
| Error | r x f x (m – 1) | \= 680 |
| Total | r x f x m – 1 | \= 815 |
The above ANOVA table looks very similar to the one from our incorrect analyses, but it incorporates an additional row for the plot factor. In this case, the significance of the replicate and family factors will be assessed using the F-values of $F\_{7,112}$ and $F\_{16,112}$. These are the same tests used for the plot-mean analysis, and they are using the correct _df_: 112 instead of 792.
The results from fitting our LMM to this dataset using our full model produces the following results:
| | Df | denDF | F.inc | Pr |
| --- | --- | --- | --- | --- |
| (Intercept) | 1 | 107.5 | 1.537e+04 | 0.0000000 |
| rep | 7 | 107.8 | 8.001e-01 | 0.5889034 |
| family | 16 | 109.2 | 1.714e+00 | 0.0541757 |
\<br/>
| family | predicted.value | std.error | status |
| --- | --- | --- | --- |
| 18 | 24.06071 | 0.7860554 | Estimable |
| 28 | 25.73520 | 0.8441194 | Estimable |
| 102 | 23.43381 | 0.8127377 | Estimable |
| 104 | 22.93619 | 0.8313801 | Estimable |
| 158 | 26.26424 | 0.7860553 | Estimable |
| 172 | 24.60833 | 0.7678512 | Estimable |
The first thing to note is that the denominator _df_ for replicate and family is ~108 instead of 112. This value is approximated using the Kenward-Roger correction for _df_, which aims to help with testing the significance of a model factor. This correction considers that under LMM often the null distribution of the test is unknown, and it accounts (penalizes) for the additional uncertainty introduced by the fact that we are using estimates of the variance component and not its true values. For this reason, the denominator _df_ approximated under Kenward-Roger correction tends to be lower than the ones expected from our ANOVA table.
For this analysis, based on a significance level of 1%, we have a p-value for the family term of 0.054, a value that will lead us to similar conclusions as the one from the plot-level data (p-value of 0.043). However, the p-value here is slightly larger because the Kenward-Roger correction has been used.
The above output resulted in predicted values for each family that are similar to the other analyses; however, the SEMs are much closer to the ones from the plot-level analysis. Again, differences are mostly due to some dead trees (_i.e._, missing data).
One additional piece of information that we can obtain from our correct LMM are the estimates of two variance components. These are obtained with the following command:
```plaintext
summary(modCC)$varcomp
```
resulting in the output:
| | component | std.error | z.ratio | bound | %ch |
| --- | --- | --- | --- | --- | --- |
| rep:plot | 1.45570 | 0.731542 | 1.989907 | P | 0 |
| units!R | 19.56639 | 1.152809 | 16.972794 | P | 0 |
The plot variance estimate $\\hat{\\sigma}^2\_p$, which corresponds to the between-plot variance, has an estimated value of 1.456. For this dataset, the estimated between-plot variance is much smaller than the residual or within-plot variance estimate, $\\hat{\\sigma}^2\_e$, with a value of 19.57. These estimates tell us that this experiment has considerably more variability between trees in a given plot (approximately 93%), than between plots. This information can be useful to improve the design of future experiments. For example, given that in this case most of the variability is at the tree-within-plot level, it might be recommended to have larger (_i.e._, more than six trees) but fewer plots than used at the present. But this might require a reduction in the number of EU if we want to keep the total number of trees constant.
It is also interesting to note that the plot-level residual variance corresponded to 5.54, a smaller value than the one obtained from our LMM, with a value of 19.57. This difference occurs because the residual variances represent different units: the former is from plot-means, and the latter is from individual plants. It is possible to connect them using the expression:
$$
\\sigma^2\_{e.plot} = \\sigma^2\_p + \\sigma^2\_{e.plant}/m
$$
where $\\sigma^2\_{e.plot}$ and $\\sigma^2\_{e.plant}$ are the residual variances for the plot-level and plant-level analyses, respectively; and $\\sigma^2\_p$ is the plot variance from the plant-level analysis. Finally, mm is the number of MU per EU. In our experiment this $\\texttt{m}$ will correspond to 6 trees/plot, but given that we have some missing values we must estimate $\\texttt{m}$ using the harmonic mean on the number of non-missing records per plot. This corresponds to $\\hat{m}$ = 4.89. Therefore, we obtain:
$$
\\hat{\\sigma}^2\_p + \\hat{\\sigma}^2\_{e.plant}/m = 1.46 + 19.57/4.89 = 5.46
$$
a value quite close to the original estimate of $\\hat{\\sigma}^2\_{e.plot}$ = 5.54.
It is possible to assess different experimental designs with varying numbers of plants per plot mm, with the use of the above equation, followed by power calculations of the statistical models. This is an additional advantage of LMM: they not only provide the correct analysis for any multi-stratum structure, but they also provide estimates of the variance components for each stratum, allowing us to understand the partition of variability of the response variable of interest.
## Closing Remarks
We have explored the consequences of pseudo-replication in some statistical models, in that they can lead to incorrect conclusions. In addition, we have fitted correct models using plot-level or plant-level data, which both led us to the expected ANOVA tables and _df_. But, which of these two is a better approach? Clearly the answer is the LMM as it uses all data at its appropriate level and also it provides us with an estimation of the contribution of the variability for each stratum. One additional advantage, which was not discussed above, is that the LMM allows us to treat missing data at random easily. In our example, there were a different number of live plants per plot. This was not an issue for our LMM methodology as it used all raw information with its appropriate weight to obtain unbiased family mean estimates.
Finally, note that our approach to deal with pseudo-replication, and indirectly with multi-stratum structure, using LMM can be easily extended to other more complex cases. For example, those where we have sub-sampling within measurement units. In all cases, using random effects that identify each stratum will take care of the complex nature of our experiment providing us with the correct degrees of freedom and therefore, our main goal of obtaining the correct statistical inference is achieved.
### **Author**
Salvador A. Gezan
### **References**
Alves, RS; de Carvalho Rocha, JRdAS; Teodoro, PE; de Resende, MDV; Henriques, EP; Silva, LA. 2018. Multiple-trait BLUP: a suitable strategy for genetic selection of _Eucalyptus_. Tree Genetics & Genomes 14, 77 (2018).
Welham, SJ; Gezan, SA; Clark, SJ; Mead, A. 2014. Statistical Methods in Biology: Design and Analysis of Experiments and Regression. Chapman and Hall/CTC.
**Notes**: SAG April-2020.
' fill='none' stroke='%23304560' stroke-miterlimit='10' stroke-width='2'/%3e%3c/svg%3e)